

A raised glucose measurement on a random blood glucose test is a red flag that warrants further investigation, especially if there is a history of diabetes in the immediate or extended family. In situations where the glucose level is high and other classic symptoms, such as polyuria, polydipsia and weight loss are present, diagnosing diabetes is not difficult. If the glucose reading is borderline, however, and the individual has no classic symptoms, diagnosis is more challenging. This article presents a case of a young boy and his diagnosis following recurrent episodes of hyperglycaemia.
Case study
Hospital presentation
JO is a 7-year-old Caucasian boy with a history of several admissions to hospital for different reasons, including urinary tract infection, tonsillitis and surgical procedures. He presented to his GP with mild nocturia. When routine tests were performed, his blood glucose results were always slightly elevated (6.6, 6.7, 6.9 and 7.4 mmol/L). When he attended hospital, his parents reported that he had recently been experiencing fatigue, as well as intermittent bed wetting, but the typical symptoms of diabetes (polyuria, polydipsia or weight loss) were not present.
JO’s weight was in the 91st percentile, at 30.5 kg, and his height was in the 75th percentile at 128 cm. He had a BMI of 18.6 kg/m2 and blood pressure of 113/69 mmHg.
Medical history
JO is the only child in the family. His birth was uneventful (full term, 2.9 kg) and there was no maternal history of gestational diabetes. He has asthma and uses a steroid inhaler (fluticasone propionate 125 µg twice daily), but it was decided that his dose was not high enough to contribute to his intermittent hyperglycaemia. He also used non-steroidal medication (montelukast, 4 mg daily) as a prophylactic measure. He had undergone left thumb surgery and recent bilateral orchidopexy. During the procedures, his glucose levels ranged from 6.6–7.4 mmol/L.
Family history
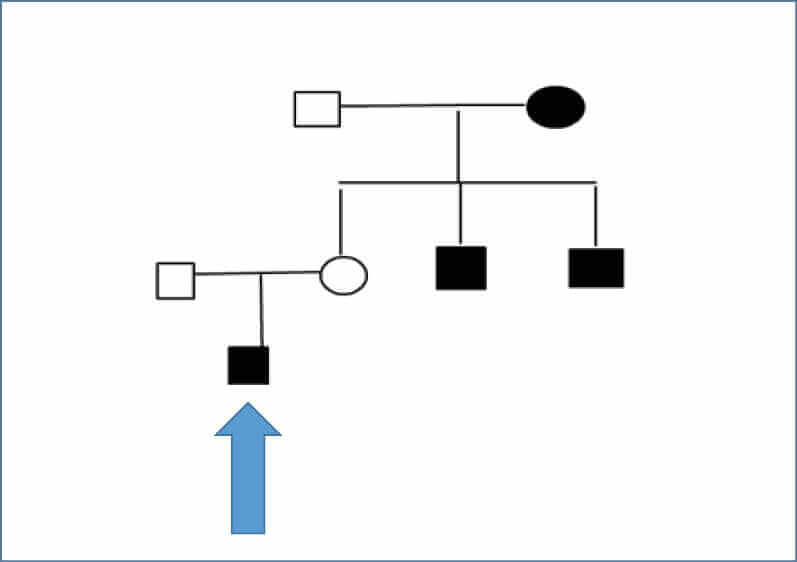
In JO’s extended family, two uncles on his maternal side had been diagnosed with type 1 diabetes at ages 11 and 13 years. In addition, his maternal grandmother has type 2 diabetes and Graves’ disease, and one of her nephews was diagnosed with type 1 diabetes at 11 years of age (see Figure 1, overleaf).
Management and progress
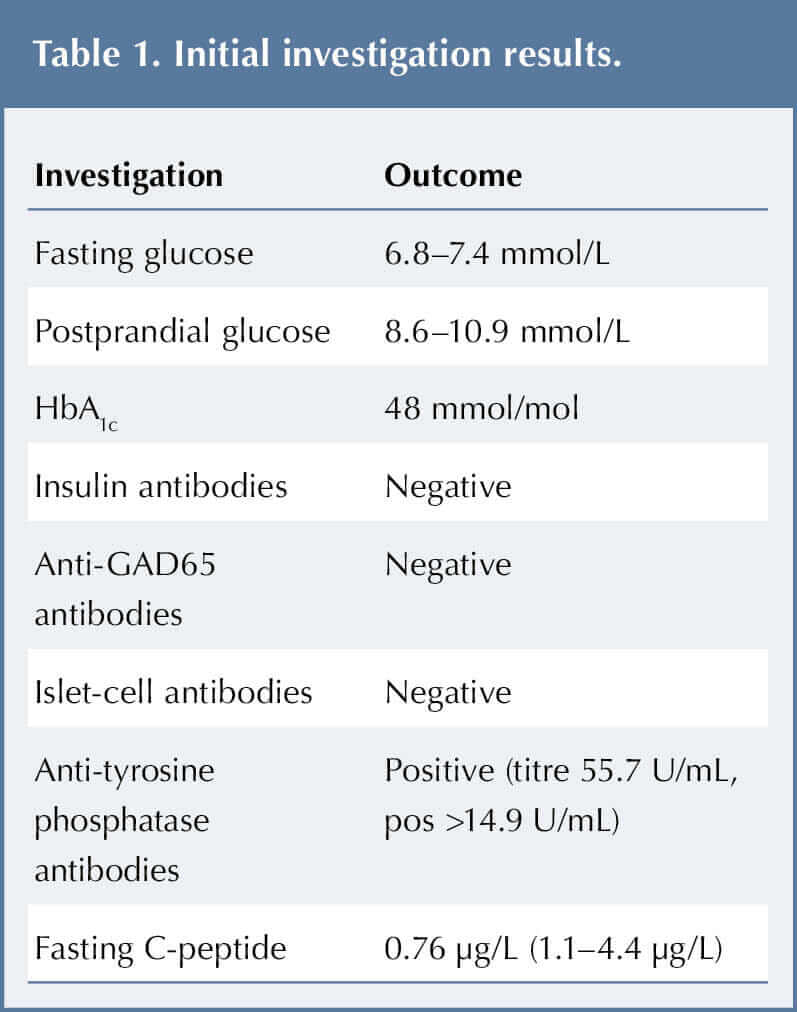
JO was admitted for further tests, including fasting and postprandial glucose, HbA1c and autoantibodies, to explore the hyperglycaemia. The results are shown in Table 1.
His initial fasting serum glucose was 6.8 mmol/L, and his 2-hour post-meal serum glucose was 7.4 mmol/L. His HbA1c was 48 mmol (6.5%). These borderline readings were not diagnostic of diabetes according to World Health Organization (2019) criteria; rather, they resembled impaired fasting glucose (IFG) and pre-diabetes.
The medical team decided to explore dietary management first. The dietitian met with JO and his mother to discuss diet and lifestyle changes to improve blood glucose control. In consultation, JO’s mother and the healthcare team agreed a plan to carry out glucose checks (fasting and 2 hours post-evening meal) at home for three days. JO’s pre-breakfast blood glucose ranged from 6.8–7.4 mmol/L, his pre-lunch from 7.0–10.3 mmol/L, and pre-dinner from 7.0–10.0 mmol/L, with mean of 8.0 mmol/L (most consistent reading). The post-evening meal glucose ranged from 8.6–10.9 mmol/L (mean 9.5 mmol/L).
Another agreed plan was to restrict and maintain JO’s fluid intake to ≤1.6 L daily, to rule out polyuria/nocturia secondary to any water-drinking behaviour. His mother followed this strictly for 3–4 weeks while continuing daily recordings in the blood glucose diary. No excessive polyuria or nocturia was reported.
Diagnosis
Diagnosis was challenging as JO tested positive for anti-tyrosine phosphatase antibody (IA-2), yet negative for other antibodies (Table 1). He also had a low C-peptide level. With the exception of some tiredness, he was asymptomatic and ketones were never identified in his urine.
In view of the above, JO’s blood was sent to the Genetic Service at Royal Devon and Exeter Hospital to test for monogenic diabetes. This confirmed a heterozygous frame shift mutation p.G285fs (c.852del) in the glucokinase (GCK) gene (glucosidase subtype), indicative of maturity-onset diabetes of the young (MODY) type 2.
JO’s autoantibody tests were also repeated about 6 months later. Surprisingly, they were negative for both IA-2 and anti-GAD65 antibodies.
Discussion
Diabetes classification
In the UK, recent figures for 2019 show there were an estimated 36,000 children with diabetes under the age of 19 years, up from 31,500 in 2015 (Royal College of Paediatrics and Child Health, 2019). The National Paediatric Diabetes Audit 2018/19 stated that the prevalence of type 1 diabetes in children and young people aged 0 to 15 years old in England and Wales was 196.4 per 100,000 of the general population; it was slightly higher among boys (198.1 per 100000) compared with girls (194.6 per 100,000).
Diabetes can be classified into the following general categories:
- Type 1 diabetes. Due to autoimmune beta-cell destruction, usually leading to absolute insulin deficiency.
- Type 2 diabetes. Due to a progressive loss of adequate beta-cell insulin secretion, frequently with the background of insulin resistance.
- Gestational diabetes. Diagnosed in the second or third trimester of pregnancy).
- Specific types of diabetes due to other causes. For example, monogenic diabetes syndromes (such as neonatal diabetes and MODY); diseases of the exocrine pancreas (such as cystic fibrosis and pancreatitis); and drug- or chemical-induced diabetes (American Diabetes Association, 2020).
Type 1 diabetes can be classified into two main types, immune-mediated or type 1A, as well as the uncommon idiopathic or type 1B, which accounts for approximately 10% of people). In contrast to type 1A, type 1B is not associated with islet cell antibodies or human leukocyte antigen, although both forms can involve ketoacidosis (DIAMOND Project Group, 2006).
Monogenic diabetes
Monogenic diabetes is uncommon, accounting for around 1–4% of all diabetes in young people (Mayer-Davis et al, 2018). It results from dominant, recessive or de novo mutations in a single gene. Abnormalities at six genetic loci on different chromosomes have been recognised and more than 40 different genetic subtypes of monogenic diabetes have been identified to date (Hattersley et al, 2018).
Maturity-onset diabetes of the young
MODY is seen in children and young adults with autosomal-dominant inherited diabetes, who have at least one family member diagnosed before 25 years of age and are negative for islet autoantibodies and acanthosis nigricans. It is characterised by a young age at onset and typically does not require insulin for at least two years after diagnosis. The child will have moderate, albeit reduced, circulating C-peptide levels 5 years after diagnosis (Winter, 2003).
Other conditions that warrant monogenic investigation include neonatal diabetes and diabetes associated with additional features, such as deafness.
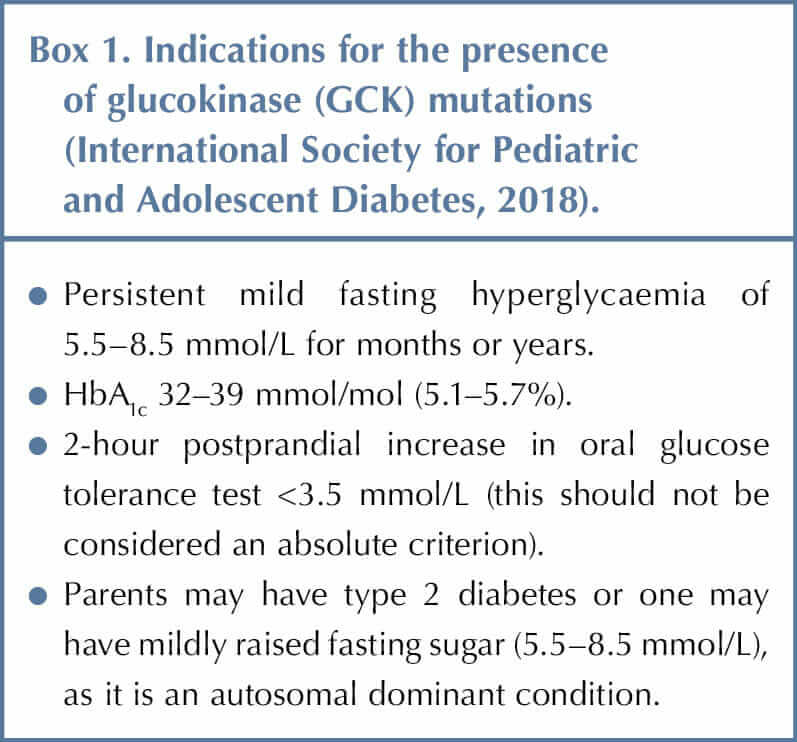
Children and young people with mild, stable fasting hyperglycaemia that does not progress should be tested for GCK gene mutations, which are the most common cause of persistent, incidental hyperglycaemia in the paediatric population (Box 1). It is also important to test the parents if considering a diagnosis of GCK mutation (Stride et al, 2002).
The population prevalence of GCK mutations is about 1.1 in 1000 (Chakera et al, 2014). The 2011 Office for National Statistics census estimated a prevalence of about 1 in 81000 of the population under 19 years of age (Uday et al, 2014).
Mild fasting hyperglycaemia due to GCK deficiency is not usually progressive during childhood. It does not need treatment and there is very little response to either oral hypoglycaemic medications or insulin. It is rarely associated with micro- or macrovascular complications, even when treatment is given throughout life (Velho et al, 1997).
Conclusions
The clinical presentation and initial investigations of this case, including low C-peptide and positive autoantibodies, were suggestive of a type 1 diabetes diagnosis and led to delayed investigations for monogenic diabetes. It is now understood, however, that autoantibodies can temporarily be positive at a young age (Vavrinec et al, 2002). In addition, the sensitivity and specificity of the assay used and laboratory cut-off values should be considered.
A formal oral glucose tolerance test was not performed in this scenario, and therefore fasting and postprandial blood results were relied upon, which was a drawback in diagnosis. The presence of persistent mild hyperglycaemia, however, warranted testing for a different cause. Consistent follow-up in such cases, in the form of twice-daily blood tests (fasting and after meals), is important. These individuals should be reviewed every 6 months and monitoring should include an HbA1c measurement. It is also prudent to repeat the C-peptide test at 12 months to monitor any decline.
Unaffected family members may initially be tested by measuring their fasting blood glucose; if elevated (>5.5 mmol/L), molecular genetic testing can then be performed.
The diagnosis of JO’s maternal uncles, who were diagnosed with type 1 diabetes at a young age, should be reviewed in the light of JO’s diagnosis. JO will require information regarding the future, as his children will have a 50% risk of inheriting this mutation and having fasting hyperglycaemia.


NHSEI National Clinical Lead for Diabetes in Children and Young People, Fulya Mehta, outlines the areas of focus for improving paediatric diabetes care.
16 Nov 2022